Introduction
In this section, we provide an overview of the data management challenges specific to single-cell sequencing experiments. Single-cell sequencing enables the analysis of gene expression at the individual cell level, leading to unique data management requirements due to the high dimensionality and complexity of the data. Single-cell sequencing encompasses a diverse array of techniques to sequence transcriptomics, epigenomics and combinations of multiple modalities (multiomics) at single-cell resolution, each presenting its own set of challenges and considerations. Additionally, the field embraces a wide range of data analysis approaches, further compounding the complexity. Consequently, addressing the standardised description and storage of data and associated metadata becomes paramount in this context. To ensure the reproducibility and reliability of research findings, it is crucial to proactively identify the specific steps in the data workflow that should be preserved. Moreover, decisions regarding data formats must be made collectively to facilitate seamless data sharing, collaboration, and long-term data preservation within the single-cell user community. This page aims to explain these challenges and provide practical guidance to navigate them effectively.
Preprocessing and quality control
Data preprocessing and quality control are integral components of the single-cell data analysis workflow, playing a pivotal role in ensuring the integrity of the data and facilitating accurate downstream analysis. These challenges encompass a spectrum of tasks aimed at enhancing data quality, reliability, and interpretation that help to make tasks conducive for subsequent analysis. By comprehensively addressing data preprocessing and quality control, we aim to provide researchers with a robust framework for navigating these critical stages of the single-cell sequencing process. This includes strategies for addressing technical variability, identifying and mitigating low-quality cells or outliers, managing batch effects, and other sources of variability that may arise within and between datasets.
Data analysis rational
Description
Preprocessing encompasses tasks such as the removal of empty droplets, quality control, batch correction, data normalisation, and transformation to mitigate technical variations. These steps aim to ensure that the data is in a suitable state for downstream analysis. Then, the next step’s central objectives include the identification of individual cells within the dataset, the assignment of gene expression profiles to each cell, and the generation of count matrices that represent the expression levels of thousands of genes across all cells. To do so, tools like CellRanger (for 10x data) or STARsolo (a more generic and open-source tool that supports various droplet- and plate-based data) are used to facilitate the crucial process of cell and gene assignment. These tools are specifically designed to take the raw sequencing data and process it into quantifiable and interpretable information. This transformation of raw data into structured, cell-by-gene matrices is fundamental for downstream analyses, such as clustering cells by similar gene expression profiles, identifying cell types or inferring cell evolution trajectories. In essence, CellRanger and STARsolo play a pivotal role in converting large and complex sequencing data into a format that researchers can subsequently explore to extract these aforementioned biological insights. Following the cell type assignation, post-processing steps come into play. These post-processing stages involve activities like efficient clustering of cells and biologically relevant annotation of clusters. By carefully orchestrating both pre- and post-processing phases, researchers can enhance the quality, reliability, and interpretability of their single-cell sequencing data, ultimately leading to more accurate and biologically meaningful insights.
Considerations
Pre-cell assignation
- Low-Quality Cell Detection: Explore methods for identifying and removing low-quality cells or outliers from the dataset.
- Normalisation and Transformation: Determine how to effectively normalise and transform the data to account for technical variability.
Post-cell assignation
- Batch Effects Handling: Develop strategies to mitigate batch effects (cell corrlate base on technical and not biological parameter) and other sources of variability within and between datasets, usually done during quality control step.
- Efficient Clustering: Consider techniques to achieve efficient and meaningful clustering of single-cell data.
- Biological Annotation: Determine how to annotate the identified cell clusters with biologically relevant labels.
Solutions
- Normalisation and Transformation: Consider using established methods such as shifted logarithm, variance stabilizing transformation (sctransform) or cell pool-based size factor estimators (scran) to address differences in sequencing depth and monitor data quality. Alternative normalisation methods such as term frequency-inverse document frequency (TF-IDF) are well-suited for scATAC-seq data.
- Low-Quality Cell Detection: Evaluate metrics like the number of detected genes per cell, mitochondrial gene content, and Unique Molecular Identifier (UMI) counts to define quality criteria. The acceptance threshold for data quality varies depending on several factors such as the amount of replicated and/or the cell type of organism (prokaryotic, eukaryotic, etc) used.
- Batch Effects Handling: Examining your data to check that the most important elements for the clustering/cell comparison are biological and not technical. Exploring batch correction methods like Harmony can help reduce technical biases in data integration.
- Biological Annotation: Use known marker genes or reference-based annotation to assign cell types or states to clusters. A database of known cell markers (like CellMarker) can be helpful.
Each of these elements needs to be provided with a comprehensive description. Including details on the normalisation techniques applied, outlier removal strategies, and batch correction methods employed to enhance data quality and reliability.
Data integration and analysis across experiments
Description
The analysis of single-cell sequencing data frequently requires the integration and comparative examination of data stemming from various experiments. Combining datasets to gain a broader perspective or comparing results from distinct experiments, navigating the intricacies of data integration, harmonisation, and interpretation is essential for extracting meaningful insights from single-cell sequencing data. This section addresses these considerations and provides solutions to facilitate the effective analysis and interpretation of integrated data, allowing researchers to draw comprehensive conclusions from diverse experimental sources.
Considerations
- Data Integration: How to integrate data from different experiments while accounting for differences in experimental conditions? (e.g. sample control vs tested conditions with several time points)
- Data Comparison: What approaches can be used to identify shared cell types and biological signals across datasets? (eg. sample WT vs KO comparison scATAC- and scRNA-seq)
- Annotation Consistency: How should we manage metadata and annotations to ensure a consistent interpretation across experiments?
Solutions
- Data Integration and Data Comparison: Use a built-in method for data integration and comparison (such as Seurat or Scanpy), including normalisation, batch correction method and dimensionality reduction techniques to see their effect. Here the difficulty is to make sure the integration/comparison is reliable, meaning being careful that the cell type annotations are consistent with previous knowledge and that the cell repartition is relevant.
- Annotation Consistency: Consistent metadata and annotation practices are needed, including standardised naming and format usage. Re-using terms from UniProt or Gene Ontology should be considered.
Datatype consistency and interoperability across formats
Description
Single-cell sequencing data is encoded into many different competing formats, with Hierarchical Data Format (HDF5)-compatible formats such as AnnData and Loom, as well as other commonly-used formats such as Seurat, CellDataSet (CDS) and SingleCellExperiment (SCE). Each of these formats is favoured by their respective analysis suites; Scanpy, Seurat, Monocle and Scater.
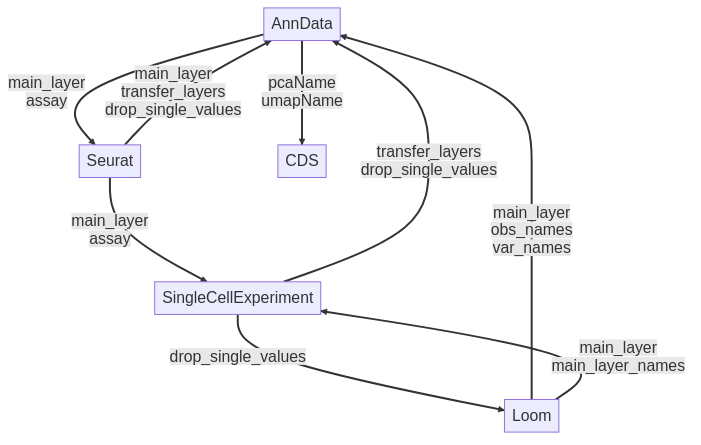
Figure 1: Conversion routes for different datatypes from sceasy
Figure 1 depicts the conversion routes of a popular conversion tool SCEasy, which demonstrates the limited conversion potential between the different formats. Indeed, data are stored in a matrix composed of different layers, converting the format may lead to the loss of some of them as described in the image. Some of these formats use different programming languages to perform the conversion, such as the Loom format which requires a Python component.
Considerations
- Datatype Preferences: Which datatypes should be actively maintained and supported, and which ones should be discouraged? Due to, for example, popularity level in the community, complexity of format, stability between versions?
- Datatype Support: Which datatypes do we actively support via bioinformatic cloud pipelines and tutorials?
Solutions
- Datatype Preferences: The most common formats are AnnData from Scanpy and SeuratObject from Seurat. There is waning support for Loom, CellDataSet and SingleCellExperiment, though SingleCellExperiment is a common datatype for sharing single-cell experiments in publications.
- Datatype Support: Seurat and Scanpy are popular analysis workflows in Galaxy, and it might be important to ensure that there is consistent and stable conversion potential between the two format (to allow the use of external tool that require the other format).
Long-term data storage and accessibility
Description
Ensuring the long-term storage and accessibility of single-cell sequencing data pose distinct challenges that demand attention. This section delves into the critical considerations for effectively storing and making single-cell sequencing data accessible over an extended period of time.
Considerations
- Effective Archiving: What are the best practices for archiving and safeguarding extensive single-cell sequencing datasets to ensure their long-term preservation?
- Ethical Data Handling: How can we guarantee data privacy and adhere to ethical guidelines when sharing confidential single-cell data with the research community?
- Collaborative Platforms: Which platforms or repositories are suitable for simplifying data sharing and encouraging collaboration among researchers?
- Enhancing Reproducibility: What specific steps and formats should be employed to enable reproducibility in single-cell sequencing experiments?
Solutions
-
Effective Archiving: Use established data repositories such as Gene Expression Omnibus (GEO) (GEO) or ArrayExpress for storage of experimental descriptive metadata and processed data such as count matrices. The corresponding raw sequencing data can be optimally archived at Sequence Read Archive or European Nucleotide Archive (ENA). Annotare can be used to facilitate deposition of data to the ArrayExpress collection in BioStudies. This tool offers support for various metadata standards; in the case of single-cell experiments it is recommended to follow the Minimum Information about a Single-Cell Experiment (minSCe) guidelines.
-
Ethical Data Handling: Emphasise the importance of informed consent and ethical considerations in data-sharing agreements. Following the privacy and ethical regulation of the hosting country or institution, using the data sharing infrastructure corresponding to the level of privacy required by the data.
-
Collaboration Platforms: Explore version control systems (e.g. Git), data sharing platforms (e.g. Zenodo), data analysis platforms (e.g. Galaxy), and domain-specific repositories (e.g. Single Cell Portal) to facilitate efficient data sharing, analysis, and collaboration.
-
Enhancing Reproducibility: Guide on enhancing reproducibility, including the use of containerisation technologies such as Docker to encapsulate analysis environments and to ensure analysis can be reproduced with the exact same tool version. Particularly, BioContainers comes in handy when dealing with bioinformatics tools. Prioritise the documentation for analysis workflows, code, and metadata using standardised formats and sharing them in version-controlled repositories. Galaxy provides a solution for containerisation, versioning, workflow management and reproducibility for novice users.
Data analysis steps and related format for single-cell sequencing
- Raw Sequencing Data:
- Data Type: Raw FASTQ files for sequencing reads.
- Format: Compressed FASTQ format (*.fastq.gz).
- Explanation: Raw sequencing data is typically stored in compressed FASTQ format (*.fastq.gz). This format retains the original sequencing reads and is space-efficient. Compressed files reduce storage requirements while preserving data integrity.
- Cell-Gene Assignment:
- Data Type: Cell-gene assignment matrix indicating gene expression levels per cell. Additionally, gene and cell annotations (e.g. gene symbols or batches, time points, genotypes) should be saved.
- Format: Standardised data matrix format, such as Hierarchical Data Format (h5), h5ad from AnnData or CSV.
- Explanation: The cell-gene assignment matrix, representing gene expression per cell, is best stored in a standardised format like Hierarchical Data Format (h5), h5ad from AnnData or CSV as it will allow the modification needed for the next step while being readable by most single-cell tools.
- Dimensionality Reduction and Clustering:
- Data Type: Reduced-dimension representations (e.g. PCA, t-SNE) and cell clusters.
- Format: Include plots and files in common data visualisation formats (e.g. PDF, PNG).
- Explanation: Visual formats like PDF and PNG allow easy sharing and visualisation.
- Annotation and Biological Interpretation:
- Data Type: Annotated cell types, differential gene expression results, and any other biologically meaningful annotations.
- Format: Structured and standardised annotation files, such as Excel spreadsheets, CSV or JSON, alongside visualisations like heatmaps or volcano plots in common visualisation formats.
- Explanation: Biologically meaningful annotations, including cell types and differential gene expression results, should be stored in structured formats. Visualisations like heatmaps or volcano plots should be included in standard visual formats for easy interpretation.
- Analysis Code and Environment:
- Data Type: All code and scripts used for data preprocessing, analysis, and visualisation.
- Format: Version-controlled repositories using Git, or container files to capture analysis environments should be used. Detailed documentation for code execution should be provided.
- Explanation: Analysis code and scripts should be version-controlled using Git repositories. Additionally, capturing the analysis environment using Docker or Singularity container files helps to ensure reproducibility. Detailed documentation of code execution is essential for transparency and re-usability.
- Metadata:
- Data Type: Comprehensive metadata describing experimental conditions, sample information, and data processing steps.
- Format: Structured metadata files in widely accepted formats like JSON, CSV or Excel spreadsheets, following community-specific metadata standards if available.
- Explanation: Following community-specific metadata standards, if available, ensures consistency and compatibility with other datasets.
By preserving these steps and data in standardised and accessible formats, researchers can enhance the reproducibility of single-cell sequencing experiments, facilitate collaboration, and ensure that others can validate and build upon their work effectively. Other additional files can be kept if useful for the interpretation (e.g. for scATAC-seq, the results files containing sequence fragments or mapping can be important, they should be kept in standardised format: tsv, bam, bed or bigwig). An overview of the best practice can be found following the Single-cell best pratices
Related pages
Galaxy is an open, web-based platform for accessible, reproducible, and transparent computational research.
The FAIRtracks ecosystem provides technical solutions for the FAIRification of genome browser track files
More information
Links to FAIRsharing
FAIRsharing is a curated, informative and educational resource on data and metadata standards, inter-related to databases and data policies.
Training
Skip tool tableTools and resources on this page
| Tool or resource | Description | Related pages | Registry |
|---|---|---|---|
| AnnData | Python package for handling annotated data matrices in memory and on disk, positioned between pandas and xarray. | Tool info Training | |
| Annotare | Annotare is a tool for submitting functional genomics data to the ArrayExpress collection in BioStudies. | Training | |
| ArrayExpress | A collection in BioStudies for archiving and publishing data from high-throughput functional genomics experiments. | Microbial biotechnology Data interlinking Data publication | Tool info Standards/Databases Training |
| BioContainers | BioContainers Flow | Data analysis | Tool info Training |
| BioStudies | A database hosting datasets from biological studies. Useful for storing or accessing life sciences data without community-accepted repositories, and for linking components of data from multi-omics studies. | Microbial biotechnology Plant sciences Data interlinking Data publication Project data managemen... | Tool info Standards/Databases Training |
| CellDataSet | The main class used by Monocle to hold single cell expression data. CellDataSet extends the basic Bioconductor ExpressionSet class. | ||
| CellMarker | A database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. | Tool info | |
| CellRanger | A set of analysis pipelines that perform sample demultiplexing, barcode processing, single cell gene counting, V(D)J transcript sequence assembly and annotation, and Feature Barcode analysis from single cell data. | Training | |
| Docker | Docker is a software for the execution of applications in virtualized environments called containers. It is linked to DockerHub, a library for sharing container images | Data analysis | Training |
| European Nucleotide Archive (ENA) | A record of sequence information scaling from raw sequcning reads to assemblies and functional annotation | Galaxy Plant Genomics Biodiversity Epitranscriptome data Human pathogen genomics Microbial biotechnology Virology Data brokering Data interlinking Data publication Project data managemen... | Tool info Standards/Databases Training |
| Galaxy | Open, web-based platform for data intensive biomedical research. Whether on the free public server or your own instance, you can perform, reproduce, and share complete analyses.
|
LabID Marine Metagenomics Virology Data analysis Data provenance Data storage | Tool info Training |
| Gene Expression Omnibus (GEO) | A repository of MIAME-compliant genomics data from arrays and high-throughput sequencing | Microbial biotechnology Data publication | Standards/Databases |
| Gene Ontology | The Gene Ontology Consortium continues to develop, maintain and use a set of structured, controlled vocabularies for the annotation of genes, gene products and sequences. | Tool info Standards/Databases Training | |
| Git | Distributed version control system designed to handle everything from small to very large projects | LabID Data organisation | Training |
| Harmony | Fast, sensitive and accurate integration of single-cell data. | Tool info Training | |
| Hierarchical Data Format | HDF5 consists of a file format for storing HDF5 data, a data model for logically organizing and accessing HDF5 data from an application, and the software (libraries, language interfaces, and tools) for working with this format. | Standards/Databases | |
| Loom | Loom is an efficient file format for large omics datasets. | Training | |
| Minimum Information about a Single-Cell Experiment (minSCe) | MinSCe is a minimum set of metadata categories used to describe a single-cell assay in sufficient detail to enable the analysis of transcriptomic data. | Standards/Databases | |
| Monocle | The Monocle 3 package provides a toolkit for analyzing single-cell gene expression experiments. | Tool info Training | |
| Scanpy | Scalable toolkit for analyzing single-cell gene expression data. It includes preprocessing, visualization, clustering, pseudotime and trajectory inference and differential expression testing. | Tool info Training | |
| Scater | Scater offers a collection of tools for analysis (such as quality control) for single-cell gene expression data. | Tool info Training | |
| SCEasy | sceasy is a package that helps easy conversion of different single-cell data formats to each other. | Tool info | |
| Sequence Read Archive | Sequence Read Archive (SRA) data, available through multiple cloud providers and NCBI servers, is the largest publicly available repository of high throughput sequencing data. | Cancer data Epitranscriptome data | Standards/Databases Training |
| Seurat | Seurat is an R package designed for QC, analysis, and exploration of single-cell RNA-seq data. | Tool info Training | |
| Single Cell Portal | Interactive portal for single-cell genomics data. | ||
| Single-cell best pratices | An entry point for novices in the field of single-cell (multi-)omic analysis and guides advanced users to the most recent best practices. | ||
| SingleCellExperiment | The SingleCellExperiment class is a lightweight Bioconductor container for storing and manipulating single-cell genomics data. | Tool info | |
| STARsolo | A comprehensive turnkey solution for quantifying gene expression in single-cell/nucleus RNA-seq data, built into RNA-seq aligner STAR. | Training | |
| UniProt | Comprehensive resource for protein sequence and annotation data | Galaxy Enzymology and biocata... Intrinsically disorder... Proteomics Structural Bioinformatics Machine actionability | Tool info Standards/Databases Training |
| Zenodo | Generalist research data repository built and developed by OpenAIRE and CERN | FAIRtracks LabID Plant Phenomics Bioimaging data Biomolecular simulatio... Enzymology and biocata... Plant sciences Data publication Identifiers | Standards/Databases Training |

Johan Rollin
University of Freiburg / MeInBio program

Mehmet Tekman
University of Freiburg / European Galaxy team

Pavankumar Videm
University of Freiburg / European Galaxy team

Bert Droesbeke
VIB Data Core / ELIXIR-BE

Federico Bianchini
University of Oslo / ELIXIR-NO

Nazeefa Fatima
Centre for Bioinformatics, University of Oslo / ELIXIR-NO